
Home / Journal / Cancer Studies
From Driver Mutations To Driver Cancer Networks: Why We Need A New Paradigm
BRAF network hub complex systems
Nikolaos I. Kalosidis, Aegli Mantsou and Nikolaos A. Papanikolaou
DOI: 10.31532/CancerStud.2.1.001 30 Mar 2018
Download

Abstract
Understanding the biochemical mechanisms of tumorigenesis remains a challenge. This is due to the hierarchical organization of cells and tissues which complicates the linking of genomic events, such as mutations, to phenotypes. Biomolecules function in complexes and in networks whose properties depend on signaling, mutation status, location and association with other proteins. In turn, interacting complexes, which are mediated by protein-protein interactions, form structural and functional modules that are part of signaling cascades, leading to the formation of networks that govern cell phenotypes. Protein-protein interactions can be modified by mutations or by different combinations of interacting surfaces. Therefore, understanding the principles of protein complex and network formation, and the effects of mutations on them, is crucial to understanding tumorigenesis. In this work we further develop the hypothesis that, besides hub proteins (proteins with the most functional/physical links in a network), we need to identify, alternate interactors resulting from mutations or altered signaling proteins in intracellular networks and intercellular networks, controlling tumor growth. We postulate that a key driver of phenotypic switching in tumor development is the formation of alternative complexes by oncogenic proteins, via network shortest paths, which control rate-limiting steps. Rate-limiting steps are the only ones that can increase pathway activity and this requires the application of complex systems dynamics analysis. Oncogene addiction-driven diversion of oncogenic signaling in mutated/overexpressed kinases in melanoma tumorigenic pathways is used in support of our proposal.
Keywords
BRAF; network; hub; complex systems
Introduction
Discovering general principles that govern the formation of alternative tumor protein complexes and networks is important for controlling growth. Previously we have argued that in order to understand tumor behavior we need to accomplish the following: First, we must reconstruct intracellular and intercellular tumor networks, and second, identify hubs that may control their behavior.1 Hubs are defined as proteins that have the most network links and tend to be crucial for network activity.2,3 Third, we must discover interactors of oncogene hubs and understand how the expression of mutated/overexpressed proteins, such as for example BRAF, EGFR or CDK4, change the interactions and links to other networks.4,5 Lastly, in terms of protein structure and conformation, we need to establish common principles that govern alternative signaling within and between networks by way of alternative hub physical connectivity.
A prominent characteristic of cellular signaling complexes is that they are chiefly enriched in disordered proteins. 6 Intrinsically disordered proteins, IDPs, and disordered regions, contain short linear motifs (SLiMs) which are 3 to 10 amino acids long. These regions mediate protein interactions endowing proteins with flexibility. Therefore, it is reasonable to hypothesize that changes in protein conformation resulting from oncogenic driver mutations or from oncogenic protein overexpression, as in oncogenic kinases such as BRAF in melanoma, lead to altered interactions within as well as between different signaling complexes thus switching signaling to alternative pathways. This feature permits phenotypic switching in polarity complexes during the epithelial-to-mesenchymal transition in invasive cancer. 7 We propose that, also it may be the case in phenotypic switching of drug-addicted melanoma cells and a general strategy of cancer cell survival.8 Consequently, altered architectures of signaling complexes in tumors may relay survival signals through different known or novel network nodes, exploiting their flexibility, probably via disordered regions. How mutation-driven, novel interactions assume full control over a network, remains an open issue,9 but whatever the case, the novel pathway(s) must be rate-limiting if they are to affect network control and pathway activity.10
Identifying and cataloguing mutations is the first step Efforts to unravel genomic changes that drive cancer growth have focused mainly on identifying driver mutations in oncogenes,11,12 have revealed complex molecular portraits of tumor heterogeneity and consequently of tumor robustness.13 Notably, different samples from the same type of tumor or the same patient, carry their own distinct cancer gene mutational signature. 13-15 This heterogeneity partly explains the lack of complete responses to targeted therapy in most patients.16,17 Our hypothesis predicts that tumors of uniformly classified histology and grade are controlled by different protein networks which, nevertheless, control similar biological functional modules such as proliferation, migration, invasion and angiogenesis. It is our hypothesis that they are driven by alterations in key complexes of mutated, as well as non-mutated proteins. Evidence exists to support this contention: First, the sequencing data reveal that in tumors of the same type, oncogenic signaling can be subsumed by different sets of active, but not necessarily mutated, kinases even if the discovered kinases, within the same family of paralogs, are mutated. Frequently, the kinases are mutated at different positions and are found in tumor cell subpopulations. As discussed before, these data reflect the heterogeneity in cell types that comprise tumors, since most studies examine mixtures of tumor cells, and the altered tumor microenvironments such as the autocrine/paracrine cytokine networks that are driven by different expression profiles compared to normal ones even if they carry no mutations themselves. 18 They are thus molecularly heterogeneous and they are therefore governed by different driver networks which we propose are driven by altered complexes. It is those driver networks and their constituent complexes that we need to identify both within tumor cells and between tumor and “healthy” tissues.19 This picture is reflected in the poor response to chemotherapeutics and to targeted biological therapies. For example, only 7% of NSCLC patients have EGFR mutations and of these only 50% respond to Iressa with no increase in survival. Stephens and his colleagues sequenced 72 breast tumors and 9 cell lines for mutations in kinases and established that of 518 kinases only 6 had two non-synonymous mutations far below the 5% cutoff point.15 Similar results have been obtained for lung12 and testicular cancer.11 Global analysis of mutations in exomes and phosphoproteomes from ovarian cancer cells lines, with computational methods, has identified network-attacking mutations (NAMs) which re-wire signaling and, notably, shift it towards phosphothreonine.20 Although these studies are significant and shed light on activated cancer networks, they are not focused on identifying either the hubs or their novel interactions in these networks. Moreover, since gross tumor samples are used, it is not clear to which type of tumor cell the networks belong. These issues are crucial if we are to successfully target tumor tissue robustness. Our hypothesis is also supported by the study of Drake and colleagues who found that different cancer hallmarks and different networks are seen in samples from five patients when they used mutational information only. Using phospho-based activities of tyrosine kinases together with mutational data, they found that the phospho-based activities were as predictive of drug responses as mutational status alone, suggesting that either one could be used to infer pathology. However, different patients with the same tumor exhibit different networks, even in the absence of mutations,21 suggesting that single and even multiple-agent approaches will fail.
What are the implications of the above findings for rational targeting? The answer to these questions seems to be that the same biological modules (or hallmarks) of function that characterize tumors such as invasion, proliferation and survival, can be controlled by redundant systems of co-expressed gene products organized in different networks, populated by different multiprotein complexes, different PTM combinations, and driven by heterogeneous mutations in DNA. We hypothesize that when a key protein kinase or transcription factor such as for example BRAF or MYC is mutated, it leads to the formation of different protein complexes by altered associations, driven by changes in protein conformation. Altered interactions due to altered conditions (e.g. overexpression of an oncogene) can be mediated by intrinsically disordered regions, IDRs, whose flexibility permits key proteins to participate in different signaling complexes. Such changes are likely to place enzymes, such as kinases and methylases, in a different context that relays aberrant signaling through a different network.22 Alternative complex formation is likely to be driven by proteins with disordered regions which are characterized by the presence of short linear motifs, SLiMs.23 Since disordered proteins are critically involved in signaling, we suggest that SLiM-driven, oncogenically altered interactions are likely to be responsible for phenotypic switching and for tumor behavior.6
Mutations, protein complexes and genetic heterogeneity Tumors are complex communities of different cells with different mutation signatures and different properties; they are tissues growing at the wrong place and time. At the molecular level, this is reflected in the extraordinary diversity and number of mutations at the genome level.14,24,25 It is, however, unknown how mutated proteins, especially hub proteins, contribute mechanistically to the biological behavior of tumors via altered interactions. We are confronted with the fact that our genetic and genomic discoveries are having only a minor effect on efforts to control cancer in the clinic. We argue that in order to link genomic alterations with tumor mass behavior, we must first understand the architecture of tumors, i.e. the types of cells and second the networks that sustain this architecture. Phenotypic switching via altered hub interactions may be a key mechanism that endows tumors with robustness. Therefore, we must define the links between the intracellular and intercellular networks that promote functional interrelationships that drive tumor growth in a systems-wide manner (Figure 1) and also that genetic interaction data should be correlated with functional protein complexes.26 Although many types of tumors express fusion oncogenic proteins, evolutionarily new phenotypes arise mainly through novel connectivities between existing or modified (e.g. mutated) proteins in multiprotein complexes, and rarely from the generation of novel proteins. Thus it is essential to define alterations in complexes formed by mutated proteins as well as their normal counterparts.27,28 Existing experimental evidence indicates that the rewiring of novel connectivities of mutated or overexpressed oncogenic proteins, acting as hubs with novel partners in intracellular or intercellular networks, may be a key mechanism of tumor survival (Figures 1 and 2). It is, therefore, imperative that we identify how such rewiring occurs. We propose that, in addition to identifying hubs, non-hub proteins may be implicated as alternative interactors however they must be part of rate-limiting steps in the signaling pathway that has assumed cell survival in order for them to be consequential.
Figure 1. Schematic representation of the hierarchical organization of information flow from complexes to phenotypes
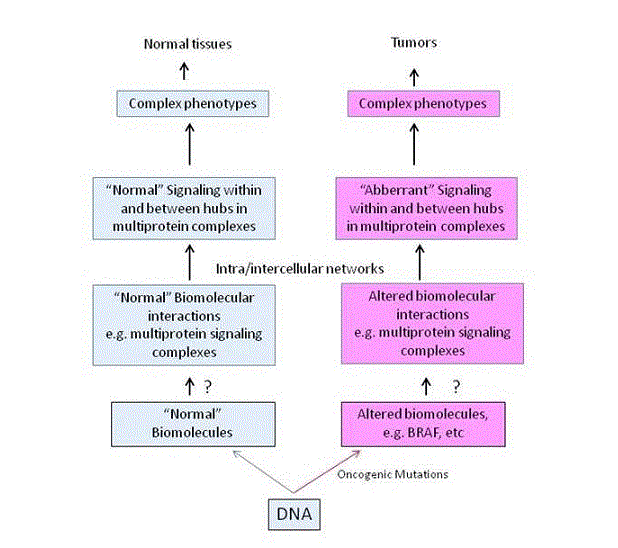
Information flow is from normal (left, blue panel) or mutated proteins (right, red panel) to multiprotein complexes, to networks and to complex phenotypes. Question marks indicate the lack of knowledge of how mutated proteins, such as for example BRAF, form altered complexes and how signaling is diverted to other networks that support growth or drug addiction of tumor cells.
Figure 2. Graphical representation of intersection of BRAF, ERK1, ERK2, MEK and MITF sub-network interactors extracted from the Pickle database
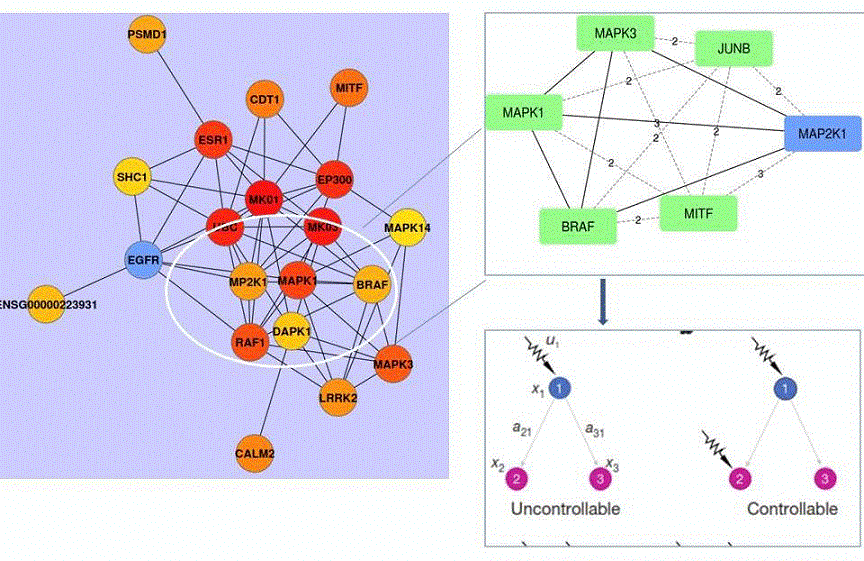
Left panel: BRAF sub-network neighborhood showing all BRAF neighbors. Ellipse contains key BRAF neighbors that are involved in melanoma cell addiction/resistance to target BRAF and MEK (MAP2K1). BRAF interacts directly with ERK2 (MAPK1) or ERK1 (MAPK3) and with MEK (MAP2K1). Nodes are round balls and edges, representing interactions or genetic links, are black lines linking nodes. Upper right panel: Shortest paths between BRAF and its neighbors are shown in broken lines. The numbers are calculated numerical values of the shortest path between two nodes. The larger the number the higher the number of edges (functional or physical links) required to functionally link the nodes. Lower panel: Controllability of three nodes receiving either a single signal (signal u1, lower left panel) or two independent signals (two arrows, lower right panel). Proteins (nodes) are shown as colored balls or squares and functional/physical links (edges) as lines.45 Node 1(x1) is BRAF, node 2 (x2) is ERK2 and node 3 (x3) is ERK1. Although they all share common interactors, such as KSR2 (Table 1), it is unknown how inhibition of BRAF or MEK diverts signaling to ERK2, but not to ERK1, to support survival of melanoma and in lung cancer cells.
Table 1. Intersection of shared interactors between BRAF, MAPK3 (ERK1), MAPK1 (ERK2), MEK, JUNB, FOSL and MITF
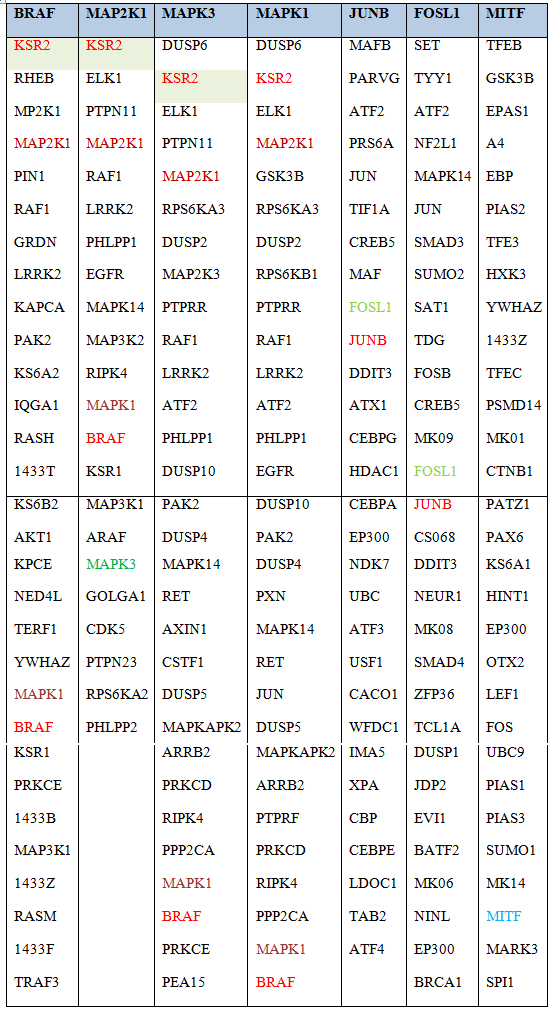
Known direct interactors of BRAF, ERK2 (MAPK1), ERK1 (MAPK3), MEK (MAP2K1), JUNB, FOSL and MITF were extracted from the database pickle.gr which integrates all experimentally verified physical interactions from the following databases: STRING (https://string-db.org/), MINT (http://mint.bio.uniroma2.it/), HPRD, (http://www.hprd.org/), BioGrid (https://thebiogrid.org/), InAct (https://www.ebi.ac.uk/intact/), and DIP (http://dip.doe-mbi.ucla.edu). Shown in red, green and brown, are proteins shared between BRAF, ERK1, ERK2 and MEK. Extraction was restricted to experimentally verified, functional or physical interactions only.
Our hypothesis is consistent with the existence of oncogene-addiction29,30 and non-oncogene addiction31,32 for the following reasons. First, altered complexes of mutated proteins rewire signaling through alternative and known signaling partners taking advantage of the fact that many proteins share interactors provided the interaction is affecting rate-limiting steps (Figure 2, Table 1). Second, through interactions with novel partners resulting from the generation of altered amino acid surfaces around mutated amino acids in disordered regions, thus relaying survival signals through another network and pathway (Figure 2). Disordered regions and short linear motifs (SLiMs) are good candidates for altered interactions since they are enriched in oncogenic signaling proteins.33 In fact, it is estimated that 40% of proteins is extensively disordered or contains IDRs, and SLiMs in disordered regions, making them flexibly adaptable for engaging in novel interactions.
Rules of engagement: From mutations to different multiprotein signaling complexes It is unknown how mutations or overexpressed oncogenic proteins, such as BRAF, rewire multiprotein signaling complexes, diverting oncogenic signals through different pathways: First, several different structure/function elements in proteins participate in altered signaling complexes that are oncogenic. Second, the reshuffling of interacting surfaces (and elements within) resulting from mutations generate novel sub-network interactions. In order for novel interactions to increase pathway activity they must affect rate-limiting steps in a pathway. Thus, when a gene is transiently expressed in normal cells or is permanently expressed in tumor cells, such as for example BRAF, or MEK in melanoma cells, it forms different complexes with different partners and substrates and can affect tumor growth at different stages.34 Such interactions may be mediated by different sequence elements such as short linear motifs (SLiMs) which interact with unknown partners under non-cancerous conditions and thus they may signal through different pathways, suggesting that catalytic and connectivity elements in proteins are reshuffled by mutant proteins to novel complexes generating new connectivities.
MAPK kinase or cyclin signaling cascade complexes exemplify the above idea since different proteins participate in different pathways while sharing some protein partners. Scaffold proteins such as cyclins or zinc-finger proteins, such as ZNF217, recruit both kinases, methylases and deacetylases and substrates to different complexes hence their expression or mutation status is critical for normal or aberrant signaling.35 For example, overexpression of ZNF217 inhibits apoptosis and increases signaling through the MAPK and PI3K/Akt pathways; however, it is unknown how ZNF217 signaling complex composition is affected by elevated levels of this scaffold/transcription factor protein in tumor cells. Data from yeast indicate that proteins distributed in different complexes have different functions and this may be a general strategy used by cancer cells. For example the yeast MAP3K kinase is active in at least three different signaling pathways; mating, invasive growth and high osmolality response whereas the hepatitis C virus protein, NS5A, although not involved in IFN-alpha signaling, interacts with several different proteins of the cellular signaling apparatus, transcription activation machinery and cell cycle-regulatory kinases, via a highly conserved C-terminal polyproline motif in a disordered region.36,37 It is unknown what happens when signaling/interaction elements in mutated proteins find themselves interacting with different proteins in alternative complexes in tumor cells and this is an area that is poorly explored. Therefore, it is reasonable to suggest that the composition of signaling protein complexes in normal vs. tumor samples where the protein is mutated can shed light on their differences and their links to novel networks.38
To support our hypothesis, we use as an example current research in melanoma and lung cancer cell lines which reveal that drug addiction results from rewired signaling, leading to phenotypic switching. Our proposal suggests that in melanoma, amplified/overexpressed BRAF engages in alternative interactions, diverting signaling to a new, cancer sustaining pathway. Also, we argue that such signaling switching must relay survival/addiction signals via rate-limiting steps and pathways. Currently, we lack knowledge of the biophysics of diverted disordered region/SLiM interactions. We speculate that the flexibility of intrinsically IDRs probably plays a role.
Figure 3. In silico analysis of intrinsically disordered regions (IDRs) of human BRAF protein (766 aa)
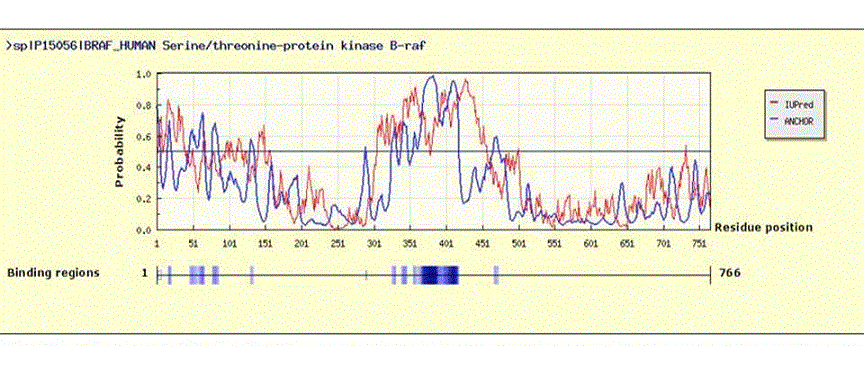
The human BRAF amino acid sequence was extracted in FASTA format from PubMed, (UniProt ID: P15056), and was used in the IuPred/ANCHOR website (http://anchor.enzim.hu/) where it was subjected to disorder analysis using default settings.48 Upper panel: Results showing disordered regions and binding regions. An amino acid sequence is considered disordered when an algorithm-generated probability of >0.5 (y-axis) is assigned. Extensive disorder is seen between amino acids 300-450 and 1-50.
The drugs dabrafenib and trametinib which target oncogenic BRAF and MEK1K57N proteins respectively in melanoma cells, induce addiction (dependence on drug presence for survival) via activated ERK2 (MAPK1), JUNB protein and the transcription factor MITF, but not via ERK1.8 These proteins share several partner proteins in their complexes (Table 1 and Figure 2) but it is unknown how a complex with overexpressed BRAF or mutated MEK links to ERK2 complexes and how this rewiring leads to drug dependence. We hypothesize that in the presence of the drugs, the mutated protein’s complexes either loose or gain novel partners which then direct signaling to a different network, perhaps that of ERK2. Although ERK1, ERK2 and BRAF share some partners (Table 1) their actual composition, when mutated, is not defined in the Kong study. In addition, it is not known how inhibiting the function of mutant BRAF or MEK in the BRAF pathway leads to ERK2 involvement in drug dependence for survival but not of ERK1. Notably, BRAF directly interacts with both ERK2 and ERK1. (Table 1). ERK1 is MAPK3 and ERK2 is MAPK1. In order to gain insight and uncover potential interactors that might explain why BRAF signaling in addicted cells is relayed via ERK2, but not ERK1, we have used BRAF, MEK, ERK1, ERK2, JUNB and MITF as queries and mined their interactors from the Pickle database which incorporates data from several different protein-protein interaction databases.39 We extracted 931 nodes having 7719 edges, a subset of which is shown in Table 1. We then constructed a BRAF network and computed its nearest neighbors (Figure 2) in Cytoscape.40 ERK1 (MAPK3), ERK2 (MAPK1), MEK (MAP2K1) and MITF are all BRAF network neighbors but not JUNB or FOSL (Figure 2, left panel). We then computed the shortest paths between BRAF and each one of its neighbors (Figure 2, right upper panel) with the cytoHubba algorithm.41 In network (graph) theory, the shortest path is the problem of finding a path between two vertices (or nodes) in a graph so that the sum of the weights of its constituent edges which may be functional or physical links is minimized. We suggest that mutant proteins generate novel shortest paths through novel interactions which represent efficient signal relays of alternative signaling in rate-limiting pathway steps. On the other hand, because static network descriptions are often inadequate at reflecting signaling reality, since they are based on simple interaction/connectivity predictions, analysis of the dynamics of complex systems and networks under different conditions may be more accurate with control theory of complex systems. According to complex systems theory, a system controlled by a set of nodes can assume full control of the network, if driven by different signals. 42,43 It is reasonable to hypothesize that this may be relevant in altered signaling interactions of oncoproteins. Kong et al. have reported that in melanoma cells addicted to BRAF or MEK inhibitors, the drug addiction pathway is relayed by ERK2, as well as by JUNB/FOSL and the transcription factor protein, MITF, but not by ERK1.8 This requires an explanation as BRAF engages in direct interactions with both ERK2 and ERK1 (Figure 2, upper right panel showing shortest paths). As predicted by complex systems controllability theory, two nodes, for example ERK2 (node 2) and ERK1 (node 3), can be controlled by a signal applied to another node such as BRAF (node 1), as depicted in Figure 2, right, lower panel. From a static network point of view, nodes 2 and 3 (ERK2 and ERK1 respectively) appear to be controlled by signal u1. However, according to the Kalman condition,44 because, as hypothesized, the signal reaches nodes 2 (ERK2) and 3 (ERK1) from a single source, node 1 (BRAF), their responses are always correlated hence they are not independently controllable, rendering the system of the three nodes uncontrollable (Figure 2, lower left panel). For all three nodes to be controllable, either node 2 (ERK2) or node 3 (ERK1) must receive an additional control signal, 45 indicated as controllable in Figure 2, lower right panel. We argue that if overexpressed BRAF engages in novel interactions, possibly via (an) as yet unknown protein(s) in rate-limiting steps, engaging ERK2 or ERK1, this protein acts as a signal relay symbolized by the arrow on ERK2 (node x2, Figure 2, lower right panel). As discussed before, typically, signaling proteins engage in interactions through SLiM motifs in disordered regions (IDRs), which upon binding, undergo a disorder-to-order transition. 46 Such binding interactions occur with low affinity and are hard to identify with large-scale screens. In support of the above, analysis of MAPK kinase docking motifs revealed novel motifs in IDRs that permit MAPKs to interact with a wide diversity of proteins in signaling thus explaining the versatility of these kinases.47 We analyzed the sequence of human BRAF for disordered regions using the ANCHOR/IuPred website 48 and we identified potential binding sites (Figure 3). An extensive disordered region exists between amino acids 300 and 450 containing predicted/verified binding sites (Figure 3, upper panel, y coordinate >0.5). Notably, binding regions fall mostly within the disordered region between amino acids 300-450, pointing in the direction of experimental validation. Furthermore, we must explain why the proposed interaction is capable of drug addiction relaying signals through ERK2 but not through ERK1 even though BRAF can interact with both kinases via the MEK kinase. A trivial explanation is that, first, the interaction with ERK1 does not increase pathway activity because it is not rate-limiting, and therefore does not alter flux through the pathway, whereas that with ERK2, being rate-limiting, increases pathway activity and therefore flux, thus switching signaling through ERK2. Second, that ERK1 and MITF have a shortest path value of 3 whereas ERK2 and MITF a value of 2, thus making ERK2 a more plausible route to relay the oncogenic signal to MITF. These hypotheses require experimental validation along with identification of alternative BRAF/ERK2 and BRAF/ERK1 interactors. These inferences also agree with the fact that static network description of connectivities representing protein interactions is insufficient to help us understand the dynamics of complex biological systems of protein complexes. Our hypothesis is experimentally testable in that normal BRAF can be expressed or overexpressed at varying levels in melanoma cells followed by isolation and characterization of its complexes. 49 Alternatively, CRISP9/CAS can be employed to introduce a tag such as the FLAG octapeptide in front of endogenous or exogenously expressed FLAG-BRAF in normal or melanoma cells that are either drug addicted or not and complexes characterized. In addition, dot-blot assays of overexpressed BRAF peptides covering its entire length but especially the disordered region from 300 to amino acid 450 from addicted and non-addicted (sensitive) melanoma cells, could shed light on how switching diverts signaling and ensures sustained tumor growth.
Conflict of Interest
The authors declare that they have no conflict of interest
Acknowledgments
This study was supported by a 2014-2015 Hopkins-Libra Foundation visiting fellowship to Dr. Papanikolaou at the Department of Biological Chemistry, John Hopkins University School of Medicine, Baltimore USA. The authors also wish to thank Dr. Barabasi of the Center for Complex Networks Research, Department of Physics, Northeastern University, Boston, Massachusetts, USA, for permission to use Figure 2a (Yan et al, 2017).We apologize to colleagues whose work we could not cite.
References
- Koutsogiannouli E, Papavassiliou AG & Papanikolaou NA. Complexity in cancer biology: is systems biology the answer? Cancer Med. 2013; 2, 164–177.
- Barabási AL & Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004; 5, 101–113.
- Yook SH, Oltvai ZN & Barabási AL. Functional and topological characterization of protein interaction networks. Proteomics. 2004; 4, 928–942.
- Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002; 62, 6997–7000.
- Li Z, Ivanov AA, Su R, et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun. 2017; 8, 14356.
- Babu MM, van der Lee R, de Groot NS & G sponer J. Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol. 2011; 21, 432–440.
- Gandalovičová A, Vomastek T, Rosel D & Brábek J. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget. 2016; 7, 25022–25049.
- Kong X, Kuilman T, Shahrabi A, et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature. 2017; 550, 270.
- Hannum, G. Srivas R, Guénolé Aude, et al. Genome-wide association data reveal a global map of genetic interactions among protein complexes. PLoS Genet. 2009; 5, e1000782.
- Dietel M. Targeted Therapies in Cancer. 2007; (Springer Science & Business Media).
- Bignell G, Smith R, Hunter C, et al. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes. Chromosomes Cancer. 2006; 45, 42–46
- Davies, H. Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417, 949–954.
- Chin SF, Wang Y, Thorne NP, et al. Using array-comparative genomic hybridization to define molecular portraits of primary breast cancers. Oncogene. 2007; 26, 1959–1970.
- Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006; 314, 268–274.
- Stephens P, Edkins S, Davies H, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005; 37, 590–592.
- Azevedo H & Moreira-Filho CA. Topological robustness analysis of protein interaction networks reveals key targets for overcoming chemotherapy resistance in glioma. Sci Rep. 2015; 5, 16830.
- Yoshida GJ & Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016; 107, 5–11.
- McDaniel SM, Rumer KK, Bioc SL, et al. Remodeling of the mammary microenvironment after lactation promotes breast tumor cell metastasis. Am J Pathol. 2006; 168, 608–620.
- Lowengrub JS, Frieboes HB, Jin F, et al. Nonlinear modelling of cancer: bridging the gap between cells and tumours. Nonlinearity. 2010; 23, R1–R9.
- Creixell P, Schoof CM, Simpson CD, et al. Kinome-wide Decoding of Network-Attacking Mutations Rewiring Cancer Signaling. Cell. 2015; 163, 202–217.
- Drake JM, Paull EO, Graham NA, et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell. 2016; 166, 1041–1054.
- Buljan, M. et al. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol Cell. 2012; 46, 871–883.
- Ren S, Yang G, He Y, et al. The conservation pattern of short linear motifs is highly correlated with the function of interacting protein domains. BMC Genomics. 2008; 9, 452.
- Bardelli A, Parsons DW, Silliman N, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003; 300, 949.
- Collier LS, Carlson CM, Ravimohan S, Dupuy AJ & Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005; 436, 272–276.
- Bandyopadhyay S, Kelley R, Krogan NJ & Ideker T. Functional maps of protein complexes from quantitative genetic interaction data. PLoS Comput Biol. 2008; 4, e1000065.
- Luck K, Sheynkman GM, Zhang I & Vidal M. Proteome-Scale Human Interactomics. Trends Biochem Sci. 2017; 42, 342–354.
- Vidal M, Cusick ME & Barabási AL. Interactome networks and human disease. Cell. 2011; 144, 986–998.
- Weinstein IB & Joe A. Oncogene addiction. Cancer Res. 2008; 68, 3077–3080; discussion 3080.
- Willis RE. Human gene control by vital oncogenes: revisiting a theoretical model and its implications for targeted cancer therapy. Int J Mol Sci. 2012; 13, 316–335.
- Luo J, Solimini NL & Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009; 136, 823–837.
- Nagel R, Semenova EA & Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016; 17, 1516–1531.
- Van Roey K, Gibson TJ & Davey NE. Motif switches: decision-making in cell regulation. Curr Opin Struct Biol. 2012; 22, 378–385.
- Dong J, Phelps RG, Qiao R, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003; 63, 3883–3885.
- Mantsou A, Koutsogiannouli E, Haitoglou C, Papavassiliou AG & Papanikolaou NA. Regulation of expression of the p21CIP1 gene by the transcription factor ZNF217 and MDM2. Biochem Cell Biol Biochim Biol Cell. 2016; 94, 560–568.
- Macdonald A, Mazaleyrat S, McCormick C. et al. Further studies on hepatitis C virus NS5A-SH3 domain interactions: identification of residues critical for binding and implications for viral RNA replication and modulation of cell signalling. J Gen Virol. 2005; 86, 1035–1044.
- Reyes GR. The nonstructural NS5A protein of hepatitis C virus: an expanding, multifunctional role in enhancing hepatitis C virus pathogenesis. J Biomed Sci. 2002; 9, 187–197.
- Cusick ME, Klitgord N, Vidal M & Hill DE. Interactome: gateway into systems biology. Hum Mol Genet. 2005; 14 Spec No. 2, R171–R181.
- Gioutlakis A, Klapa MI, & Moschonas NK. PICKLE 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PloS One. 2017; 12, e0186039.
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003;13, 2498–2504.
- Lin CY, Chin CH, Wu HH, et al. Hubba: hub objects analyzer—a framework of interactome hubs identification for network biology. Nucleic Acids Res. 2008; 36, W438–W443.
- Liu YY, Slotine JJ & Barabási AL. Controllability of complex networks. Nature. 2011; 473, 167–173.
- Liu YY, Slotine JJ & Barabási AL. Control centrality and hierarchical structure in complex networks. PloS One. 2012; 7, e44459.
- Kalman RE. Lyapunov functions for the problem of lur’e in automatic control. Proc Natl Acad Sci. U. S. A. 1963; 49, 201–205.
- Yan G, Vertes PE, Towlson EK, et al. Network control principles predict neuron function in the Caenorhabditis elegans connectome. Nature. 2017; 550, 519–523.
- Babu MM. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans. 2016; 44, 1185–1200.
- Zeke A, Bastys T, Alexa A. et al. Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol Syst Biol. 2015; 11, 837.
- Mészáros B, Simon I & Dosztányi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol. 2009; 5, e1000376.
- Westbrook TF, Stegmeier F. Elledge SJ. Dissecting cancer pathways and vulnerabilities with RNAi. Cold Spring Harb. Symp Quant Biol. 2005; 70, 435–444.