
Home / Journal / Cancer Studies
Signaling Factors Involved in Self-Renewal of Breast Cancer Stem-like Cells
Cancer stem-like cells breast cancer signaling
Kana Tominaga
DOI: 10.31532/CancerStud.2.1.003 25 Jun 2018
Download

Abstract
Since cancer stem-like cells (CSC) was identified in acute myeloid leukemia (AML) in 1997, the involvement of CSC has been reported in various cancers including glioma, breast cancer, lung cancer, intestine cancer, skin cancer, etc. Cancer cells are speculated to originate from the small CSC population; CSC also display chemotherapeutic resistance and radio-resistance. Hence, control of the CSC population may lead to the development of therapeutic strategies for inhibiting tumor growth, recurrence, or metastasis. To study CSC and their population dynamics, the flow cytometry analysis, tumor sphere culture and organoid culture would lead to the development of effective tools. Based on in vitro methods, characteristic signaling pathways in CSC were reported using these techniques, e.g., Wnt, Notch, and Hedgehog pathways. Furthermore, in silico analysis has revealed that key growth factors expressed via activation of nuclear factor κB (NF-κB) plays a role in CSC self-renewal. This review summarizes how CSC are distinguished from non-CSC and how CSC retain their self-renewal capacity through signaling or growth factors in an autocrine or paracrine manner in breast cancer.
Keywords
Cancer stem-like cells, breast cancer, signaling
Introduction
Postoperative or post-treatment survival is greater among breast cancer patients than among those with other types of cancer. However, recurrence and metastasis to the bones, lungs, and brain are possible after a latent period of 5–10 years.1 Various reports suggested that the cancer stem-like cells (CSC) are found in breast cancer and are considered to deter the prognosis of events including tumor recurrence and metastasis. 2–5 Tumors are speculated to comprise a heterogeneous cell population constituted by CSC, transit-amplifying (TA) cells, and terminally differentiated cells.6,7 CSC can undergo self-renewal, symmetric cell division, and can yield terminally differentiated cells and somatic stem cells via asymmetric cell division. 8,9 In this heterogeneous cell population, CSC, a small cell population, occupies the highest position in tumor hierarchy. With slow cell cycles and high anti-oxidative capacity compared with non-CSC, CSC are resistant to conventional chemo- and radiotherapy targeting proliferating cancer cells.10–12 Despite large cancer cell populations being eliminated through chemotherapy, only a few CSC may survive and cause tumor recurrence and metastasis. Hence, it is essential to elucidate the characteristics of CSC and standardize methods of assessing CSC-enriched populations and associated culture methods. Flow cytometry analysis using known CSC-specific antibodies is a popular and simple method for assessing CSC.13–15 The CSC population can be enriched and fractionated through flow cytometry analysis because cell membrane characteristics of CSC is analyzed in only living cells. In standardizing culture methods for CSC, tumor sphere culture and organoid culture are useful tools to assess the self-renewal capacity of CSC in vitro.16,17 Sphere culture has been used to assess the survival and self-renewal capacity of neural stem cells in culture.18 CSC-derived tumor spheres are obtained through floating cell cultures in sphere culture medium (SCM) containing neural stem cells and a cocktail of growth factors, including epidermal growth factor (EGF), basic fibroblast growth factor (bFGF) and hormones. Because CSC are resistant to loss of anchorage dependence (anoikis), only CSC can grow under the serum-free and non-adherent conditions.19,20 Hence, tumor sphere forming ability correlates with self-renewal capacity of CSC in vitro. On the other hand, organoid culture, that is three dimensional (3D) culture system, has been used as the study of cellular differentiation and morphogenesis from stem and progenitor cells containing with gut or mammary gland.21–23 Organoids are expanded in collagen gel or matrigel that based on growth medium containing EGF, TGFβ-antagonist Noggin and the Wnt-agonist R-spondin1 as growth stimuli, and ROCK inhibitor Y-27632 to avoid anoikis. 24 In CSC study, organoid culture also is a helpful method for assessing the maintenance of tumor formation because tumors are able to be reconstructed from a small number of tumor cells in vitro. 25,26 Moreover, organoid culture is able to be applied to drug screening due to obtaining many clonal organoids.27 Accordingly, organoid culture will be hopefully lead to not only assess the CSC capacity but also develop anti-CSC agents.
Controlling the CSC activity would influence the inhibition of tumor growth, recurrence, or metastasis. To establish the strategy to target CSC, it is important to identify the signaling pathways activated in CSC by using the flow cytometry and the CSC culture system. Recently, it continues to become clear that several potentials signaling pathways are activated in CSC. This review discusses signaling pathways in breast CSC and the possibilities for development of novel strategies for targeting breast CSC.
The Characteristic of CSC in Breast Cancer
Breast CSC were first reported in 2003 by Al-Hajj et al..28 They reported enrichment of breast CSC in a CD44 high/CD24low/-/Lineage- cell population (so-called CSC population) derived from clinical samples subjected to flow cytometry, and this cell population had high tumor-initiating activity when inoculated into the mammary fat pads of immunodeficient mice. After that, by using staining of the cell surface antigen and the flow cytometry analysis in a similar way, specific markers expressed in cell surface of breast CSC were found. Epithelial cell adhesion molecule (EpCAM), CD10, β1 integrin (CD29), α6 integrin (CD49f), and CD133 may be used as cell surface markers of breast CSC.28–31
Figure 1. Mechanisms of self-renewal of breast CSC through activation of Wnt, Notch, and HH pathways. Hyperactivity of multiple signal pathways retains the characteristics and self-renewal ability of breast CSC. Several antagonists of signaling pathways inhibit breast CSC self-renewal. Abbreviations: LRP5/6, low-density lipoprotein receptor-related protein 5/6; FZD, Frizzled; TCF, T cell factor; LEF, Lymphoid enhancer-binding factor; JAG, Jagged; DLL, Delta-like; NICD, Notch intracellular domain; RBPJ, Recombining binding protein suppressor of hairless; SHH, Sonic hedgehog; PTCH, Patched; SMO, Smoothened.
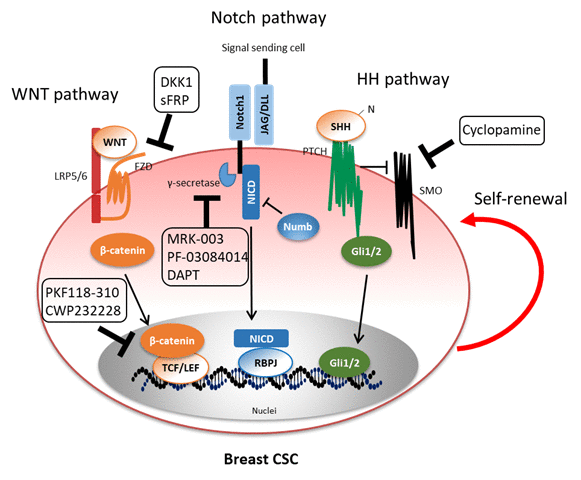
Breast CSC populations are maintained by molecules secreted from themselves (autocrine signaling) or their microenvironment, i.e., the so-called CSC niche (paracrine signaling), e.g., endothelial cells, immune cells, and cancer-associated fibroblasts (CAFs).32,33 These molecules intricately interact and regulate breast CSC self-renewal in an autocrine/paracrine manner; it has been difficult to understand distinctive signal pathways in breast CSC. However, over the past several years, potential signaling pathways activated in breast CSC have been elucidated primarily through in silico analyses. These include common pathways between cancer and somatic stem cells; however, it has been speculated that CSC are caused by abnormal and hyperactivation of various signaling pathways. Hence, we shall focus on the aberrantly activated signaling pathways, including Wnt, Notch, and Hedgehog pathways, for self-renewal of breast CSC (Figure 1). Moreover, we revealed that signaling pathways induced by key growth factors neuregulin 1(NRG) and insulin-like growth factor 2 (IGF2) to regulate self-renewal capacity of breast CSC.34,35 Inhibition of those signaling pathways by reagents may probably help develop novel therapy for eradication of breast CSC.
Key Factors for Breast CSC Maintenance
Wnt pathway The Wnt pathway regulates various functions of normal or tumor stem cells, through two pathways: canonical and non-canonical pathways.36,37 In the canonical pathway, binding of Wnt ligands to a dual receptor complex comprising the WNT co-receptors LRP5 or LRP6 and the Frizzled family (FZD1-10), a seven transmembrane domain receptor, initiates Wnt–beta-catenin signaling. The non-canonical pathway includes the planar cell polarity (PCP) pathway and Ca2+ pathway, which does not involve beta-catenin. This review refers to the canonical Wnt pathway. The Wnt pathway is involved in mammary gland development and carcinogenesis in mice or human.38,39For example,Axin2 is a direct target gene of the Wnt pathway though beta-catenin. A few of Axin2-positive stem cells generate basal and luminal alveolar cells in adult virgin mice40, while Wnt-induced Axin2 activates Snail-induced epithelial–mesenchymal transition (EMT), resulting in breast cancer cell invasion and progression41. In studies on CSC, the expression of phosphorylated beta-catenin was lower in the side population (SP), which is a flow cytometry method for detecting stem cells or CSC based on the ability to efflux the fluorescent dyes, compared with the non-SP.42When the staining intensity of cytoplasmic and nuclear beta-catenin was measured, immunostaining intensity was higher in the SP than in the non-SP, and nuclear accumulation of beta-catenin was significant in the SP. Hence, the Wnt pathway may activate in CSC. In another group, overexpression of sFRP1 or DKK1, negative regulators of the Wnt pathway, or shRNA-mediated knockdown of LRP6 reduced tumor sphere forming ability, tumor growth, and metastasis in basal-like breast cancer.43However, overexpression of Twist in immortalized human mesenchymal epithelial cells (HMLE) induced EMT and tumor sphere formation.44Because Twist, an EMT-related transcription factor, upregulatesAxin mRNA, a beta-catenin/TCF-LEF target gene, and inhibition of tumor sphere formation through treatment with recombinant sFRP1 or DKK1; this indicated that EMT-induced pathway contains beta-catenin-dependent Wnt pathway in autocrine signal and active migration and self-renewal of breast CSC. MicroRNAs (miRNAs) are important to understand the regulation of WNT signaling. Some miRNAs were reported to be regulators of CSC signaling. The high expression of miR-142 and miR-150 were observed in human breast CSC, and that miR-142 directly targetsAPC mRNA and those inducing the suppression of tumor sphere forming activity and tumor growth25. Another study reported that Let-7c expression was inversely correlated with estrogen receptor α (ERα) expression and Wnt activity.45Let-7c targets 3’UTR ofERαmRNA and inhibits breast CSC self-renewal though the APC/β-catenin/TCF pathway. Selectively targeting occurs through the Wnt pathway, e.g., PKF118-310 or CWP232228 as chemical antagonists of the Wnt pathway, inhibited breast CSC tumor sphere forming ability and tumorigenesis in a murine model.46,47Those may be candidate therapeutic agents for breast CSC maintenance through Wnt pathway
Notch Pathway The Notch pathway plays an important role in the development of breast CSC and their characteristics48. In mammals, Notch receptors comprise 4 subtypes (Notch1–4), which are activated by binding with five ligands (DLL, delta-like 1, 3, and 4; JAG, jagged 1 and 2) from signal sending cells, leading to the expression of basic helix-loop-helix (bHLH) transcription factors containing HES family or HEY family.49It is known that the Notch pathway-related genes of mammary development correlate with breast cancer cells.50 Hence, the high expression of Notch receptor correlates with poor prognosis of breast cancer patients.51In breast CSC, the Notch pathway induces hyperactivation of aldehyde dehydrogenase 1A1 (ALDH1A1), a marker of stemness, through the induction of deacetylase sirtuin 2 (SIRT2), which causes tumorigenesis and tumor growth.52Moreover, the Notch pathway in breast cancer promotes CSC self-renewal, which enhances glucose uptake and aggressive hormone-independent tumorin vivo 53; hence, the Notch pathway is associated with CSC characteristics in cancer metabolism. Recently, the Notch pathway-related proteins containing with gamma (γ)-secretase, enzyme complex related to Notch cleavage, and Notch receptors were reported to be remarkable therapeutic targets for CSC self-renewal. Treatment of MRK-003, a γ-secretase inhibitor and an antagonist of the Notch pathway, inhibit tumor sphere forming ability and the differentiation of progenitor cells.54Mice administered MRK-003 displayed apoptosis and the differentiation in ERBB2 breast cancer mouse model. PF-03084014, other γ-secretase inhibitor, used in combination with docetaxel, an anti-cancer reagent, reversed these effects and demonstrated early-stage synergistic apoptosis.55PF-03084014 used in combination with docetaxel reduces the ALDHhighand CD133+ /CD44+ CSC populations and tumor progression in the xenograft model. Other groups reported that breast cancer cells treated withN-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycinet-butyl ester (DAPT), an inhibitor of the Notch pathway, decreased the CSC population and knock-down of Notch1 or Notch4 suppressed tumor sphere forming efficiency and tumor progression.56Furthermore, brain metastatic human breast cancer cells treated with DAPT had reduced CD44high/CD24lowCSC populations compared with non-treated cells and inhibited brain metastasis in an experimental murine model.57These results suggest that the Notch pathway can regulate the characteristics of breast CSC containing self-renewal and metastatic potential.
Hedgehog Pathway The Hedgehog (HH) pathway plays a key role in various biological processes, such as cell differentiation, proliferation, and growth in normal or tumor cells.58 The HH pathway is activated by binding of ligands to the Patched (PTCH) receptor and subsequently alleviating inhibition of Smoothened (SMO).59,60 Activation of SMO results in subsequent regulation of the expression of Gli transcription factors that are responsible for cancer cell proliferation, apoptosis, and invasion. 61–64 Overexpression of molecules related with the HH pathway is observed in CSC during chronic myeloid leukemia65, medulloblastoma66, skin cancer67, etc. In breast cancer, the HH pathway activated in CSC and the mechanism underlying CSC self-renewal has been revealed recently. Using breast tumor derived from xenografts, Liu et al., first reported that the expression level of PTCH1, Gli1, and Gli2 mRNA, which are associated with the HH pathway, affects the CSC population as compared with the non-CSC population 58. Similarly, the expression level of PTCH1, SMO, Gli1, and Gli2 proteins increases in tumor spheres than in adherent cells in the breast cancer cell line MCF7. In addition, p63 is the sister homolog of p53 and a master regulator of normal epithelial stem cell maintenance 68. p63 directly regulates the expression of Shh, Gli2, and Ptch1, leading to tumor sphere formation in transgenic mice with conditional overexpression of the ErbB2 oncogene.69 Cyclopamine (CP), an antagonist of the HH pathway, is an effective inhibitor of self-renewal of breast CSC.70 Furthermore, FOXC1 binds directly to Gli2 and regulates ALDH activity and tumor sphere forming ability via activation of SMO-independent HH signaling in basal-like breast cancer. Basal-like breast cancer, lack expression of hormone receptors and ErbB2 receptor, is associated with poor prognosis in breast cancer.71 Inhibition of the FOXC1/Gli2 pathway using anti-HH inhibitors can improve basal-like breast cancer treatment.
Growth factor Signaling Pathway through Nuclear Factor-Kappa B (NF-κB)
Currently, hyperactivation of NF-κB caused by growth factor stimulation plays a key role in CSC self-renewal. NF-κB activation increases in CD24 -/Lineage- cell population compared with the other cell population, as revealed through in silico analyses using breast cancer cell lines14. Furthermore, the NF-κB pathway induced to express several inflammatory cytokines, containing Interleukin-6 (IL-6), IL-8, and CCL5 in breast cancer cells32. Because inflammatory cytokines function as paracrine factors secreted from their CSC niche cells, these observations suggest that NF-κB activation is a key factor for self-renewal of breast CSC and maintenance of those niches. Further, I introduce growth factors we have found as factors activating NF-κB in breast CSC.
Neuregulin1(NRG) NRG, also known as Heregulin (HRG), functions as a ligand of ErbB3/HER3 and activates the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway or the MEK/Erk signaling pathway.16,72 The expression of NRG is reported to correlate with clinical prognosis and chemotherapeutic resistance in several types of cancer, especially HER2-positive breast cancer. Our groups reported that NRG stimulation in breast cancer cell line MCF7 increased to form tumor spheres and expresses the stem cell marker Nanog in NRG-treated spheres; thus, NRG may be related with initiation and undifferentiation of CSC.16 Moreover, NF-κB was remarkably activated in NRG stimulation via the PI3K/Akt signaling pathway, leading to the formation of tumor spheres in primary breast tumor cells and tumor-initiating activity in xenograft models. The activated NRG/PI3K/NF-κB axis can induce IL8 mRNA expression, a regulator of self-renewal in BCSC-enriched populations. Other groups reported that NRG treatment for breast cancer cell lines increase the CD44high cell population 73 and induction of EMT through activation of Snail expression or phosphorylated Smad2 via the PI3K/Akt pathway.74 These findings suggest that NRG through the PI3K/Akt/NF-κB signaling pathway has a significant effect on the maintenance and self-renewal ability of breast CSC. Cytokines secreted owing to NRG stimulation may function as autocrine factors and paracrine factors to stimulate to CSC niche.
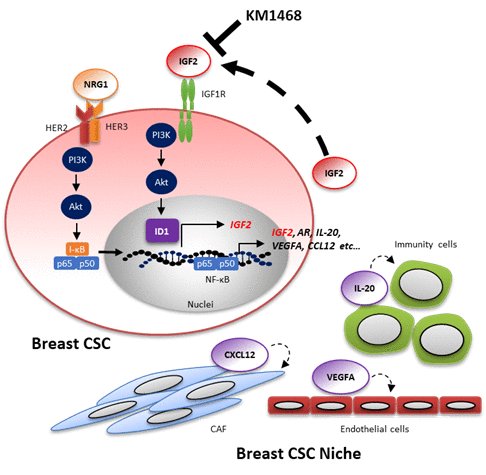
IGF2 IGF2 is a member of the insulin family and binds to IGF-1 receptor (IGF-1R) homodimers or IGF-1R and insulin receptor (IR) heterodimers. 75,76 Recent studies reported that the IGF-1R signaling pathway contributes CSC characteristics in several types of cancer. In lung adenocarcinoma, IGF-1R signaling pathway is required for chemotherapeutic resistance by altering chromatin states or acquisition of CSC characteristics.77 Similarly, IGF-1R accumulation induces the expression of FOXO3a protein (not the PI3K signaling pathway), which confers radioresistance in glioma stem cells.78 We reported that the IGF2/IGF-1R signaling pathway plays a key role in establishing tumor spheres of primary breast cancer cells34. IGF-1R was specifically upregulated in CSC-enriched populations in freshly obtained primary breast cancer cells. In addition, the IGF2/IGF-1R/PI3K signaling pathway induced the expression of the inhibitor of DNA binding protein 1 (ID1). Inhibitor of DNA-binding 1 (ID1) is a member of ID family proteins (ID1 ~ ID4) and is reported to function as master regulators for stemness of normal tissues or tumors.79 ID1 appeared to operate as a transcript regulator via upregulation of IGF2 mRNA itself, thereby probably leading to tumor sphere formation. In chemotherapy, treatment with an anti-human IGF1/2 antibody (KM1468) blocked tumorigenesis derived from the IGF-1Rhigh CSC-enriched population in a patient-derived xenograft (PDX) model. Hence, it is important that NRG1 stimulation may trigger a IGF2-ID1-IGF2 positive feedback circuit for the maintenance for stemness of breast CSC (Figure 2). On the other hand, fusion genes involved in tumor progression were recently identified in several types of tumors in lung cancer.80 The CD74-NRG1 fusion gene has been reported by several groups and its role of in lung and breast cancer has been investigated81,82; recently, the relationship between this fusion gene and CSC has been reported35. Overexpression of CD74-NRG1 in lung or breast cancer cell lines enhanced tumor sphere forming ability and tumor initiation in xenograft models. CD74-NRG1 expression promoted the expression of the secreted IGF2 and phosphorylation of its receptor, IGF-1R through the PI3K/Akt/NF-κB signaling pathway, leading to the formation of tumor spheres. This study suggests that CD74-NRG1 fusion gene expression probably contributes to CSC maintenance via the IGF2/IGF-1R signaling pathway.
Figure 2. Model of molecular mechanisms that stabilize the stemness of breast cancer cells. The HER2/HER3-PI3K-NF-κB pathway may trigger a IGF2-ID1-IGF2-mediated positive feedback circuit as a fundamental mechanism of stabilization of stemness. In addition, the HER2/HER3-PI3K-NF-κB pathway also leads to the production of many soluble factors that may regulate the surrounding CSC niche cells: cancer-associated fibroblasts (CAF), endothelial cells, immune cells, etc. KM1468 is human anti-IGF1/2 antibody and inhibits tumor initiating activity in breast cancer. This figure is adapted from Tominaga et al.34. Abbreviations: NRG1, Neuregulin1; PI3K, Phosphoinositide 3-kinase; NF-κB, nuclear factor-κB; I-κB, inhibitor of NF-κB; IGF2, Insulin growth factor 2; IGF1R, Insulin growth factor 1 receptor; ID1, inhibitor of DNA binding protein 1; AR; Amphiregulin; IL-20, Interleukin 20; VEGFA, vascular endothelial growth factor A; CCL12, Chemokine (C-C motif) ligand 12
Conclusion
This review is focused on breast CSC maintenance, including self-renewal ability through activation of signaling pathways in breast cancer. Robust autocrine or paracrine signaling, containing Wnt, Notch, Hedgehog and growth factor signaling pathways, is essential for the maintenance of breast CSC. Actually, multiple signaling pathways are complex and have cross-talk each other in cancer cells. For example, on cell lines from gastric adenocarcinomas, the expression of sFRP1 was induced by activated Hedgehog pathway, which indicated that HH pathway induced Wnt pathway inactivation83. Other group suggested that sonic hedgehog, HH pathway agonist, and Jagged 2 (JAG2), a Notch receptor, were able to reduce activated beta-catenin through SMO, leading to suppressing beta-catenin transcription in tongue cancer84. Since CSC might be thought to remain by activated or inactivated multiple signaling pathways in recurrence of breast cancer, it would be important to reveal cross-talk between signaling pathways on CSC to understand the mechanism of tumor recurrence.
In addition, growth factor-induced signaling pathways contribute to not only maintenance of breast CSC behavior, but also activation of CSC niches. Our reports using patient-derived breast cancer cells obtained from surplus surgical tumor tissues indicate that inflammatory cytokines and chemokines secreted from CSC niches regulate CSC maintenance through NF-κB activation.
To identify the specific signal pathway of CSC may probably help establish anti-cancer agents that do not affect normal tissues and somatic stem cells. Since it is possible to activate multiple and complex signaling pathways in CSC by various factors, it may be necessary to concurrently inhibit candidate targeting molecules, e.g., HER2/3, NRG, IGF-1R, IGF2, and ID1 in case of breast cancer. However, unresolved questions regarding how CSC are stimulated from their niche for the maintenance of their properties persist. Future studies are needed for novel therapeutic strategies to inhibit the bidirectional signaling pathway between CSC and their niche.
Acknowledgments
This work was supported by Grant-in-Aid for JSPS postdoc Fellows to K.Tominaga
References
- Hanrahan EO, Valero V, Gonzalez-Angulo AM, Hortobagyi GN. Prognosis and management of patients with node-negative invasive breast carcinoma that is 1 cm or smaller in size (stage 1; Tla,bN0M0): A review of the literature. J Clin Oncol. 2006;24(13):2113–2122. doi:10.1200/JCO.2005.02.8035
- Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–273. doi:10.1016/j.ccr.2007.01.013
- Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672–679. doi:10.1093/jnci/djn123
- Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;11(4):R46. doi:10.1186/bcr2333
- Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012;13(4):e178–e185. doi:10.1016/S1470-2045(11)70335-7
- Magee J a, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21(3):283–296. doi:10.1016/j.ccr.2012.03.003
- Cabrera MC. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. 2015;7(1):27. doi:10.4252/wjsc.v7.i1.27
- Roegiers F, Jan YN. Asymmetric cell division. Curr Opin Cell Biol . 2004;16(2):195–205. doi:10.1016/j.ceb.2004.02.010
- Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441(7097):1068–1074. doi:10.1038/nature04956
- Kobayashi S, Yamada-Okabe H, Suzuki M, et al. LGR5-Positive Colon Cancer Stem Cells Interconvert with Drug Resistant LGR5-Negative Cells and are Capable of Tumor Reconstitution. Stem Cells. October 2012. doi:10.1002/stem.1257
- Ishimoto T, Nagano O, Yae T, et al. CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the xCT Subunit of System xc− and Thereby Promotes Tumor Growth. Cancer Cell. 2011;19(3):387–400. doi:10.1016/j.ccr.2011.01.038
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458. doi:10.1038/nature07733
- Bleau A-M, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell. 2009;4(3):226–235. doi:10.1016/J.STEM.2009.01.007
- Murohashi M, Hinohara K, Kuroda M, et al. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br J Cancer. 2010;102(1):206–212. doi:10.1038/sj.bjc.6605468
- Li D, Su D, Xue L, Liu Y, Pang W. Establishment of pancreatic cancer stem cells by flow cytometry and their biological characteristics. Int J Clin Exp Pathol. 2015;8(9):11218–11223.
- Hinohara K, Kobayashi S, Kanauchi H, et al. ErbB receptor tyrosine kinase/NF-κB signaling controls mammosphere formation in human breast cancer. Proc Natl Acad Sci U S A. 2012;109(17):6584–6589. doi:10.1073/pnas.1113271109
- Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506–5511. http://cancerres.aacrjournals.org/content/65/13/5506.abstract.
- Reynolds BA, Rietze RL. Neural stem cells and neurospheres - Re-evaluating the relationship. Nat Methods. 2005;2(5):333–336. doi:10.1038/nmeth758
- Okabe H, Ishimoto T, Mima K, et al. CD44s signals the acquisition of the mesenchymal phenotype required for anchorage-independent cell survival in hepatocellular carcinoma. Br J Cancer. 2014;110(4):958–966. doi:10.1038/bjc.2013.759
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. doi:10.1038/nature05384
- Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009;15(6):701–706. doi:10.1038/nm.1951
- Abud HE, Watson N, Heath JK. Growth of intestinal epithelium in organ culture is dependent on EGF signalling. Exp Cell Res. 2005;303(2):252–262. doi:10.1016/j.yexcr.2004.10.006
- Shackleton M, Vaillant F, Simpson KJ, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439(7072):84–88. doi:10.1038/nature04372
- Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi:10.1038/nature09637
- Isobe T, Hisamori S, Hogan DJ, et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife. 2014;3. doi:10.7554/eLife.01977
- Merlos-Suárez A, Barriga FM, Jung P, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8(5):511–524. doi:10.1016/j.stem.2011.02.020
- Huang L, Holtzinger A, Jagan I, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med. 2015;21(11):1364–1371. doi:10.1038/nm.3973
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi:10.1073/pnas.0530291100
- Maguer-Satta V, Chapellier M, Delay E, Bachelard-Cascales E. CD10: a tool to crack the role of stem cells in breast cancer. Proc Natl Acad Sci U S A. 2011;108(49):E1264; author reply E1265. doi:10.1073/pnas.1116567108
- Lo P-K, Kanojia D, Liu X, et al. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFβ signaling. Oncogene. 2012;31(21):2614–2626. doi:10.1038/onc.2011.439
- Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar S V, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10(1):R10. doi:10.1186/bcr1855
- Hinohara K, Gotoh N. Inflammatory signaling pathways in self-renewing breast cancer stem cells. Curr Opin Pharmacol. 2010;10(6):650–654. doi:10.1016/j.coph.2010.08.003
- Korkaya H, Liu S, Wicha MS. Review series Breast cancer stem cells , cytokine networks , and the tumor microenvironment. J Clin Invest. 2011;121(10):3804–3809. doi:10.1172/JCI57099.3804
- Tominaga K, Shimamura T, Kimura N, et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene. 2017;36(9):1276–1286. doi:10.1038/onc.2016.293
- Murayama T, Nakaoku T, Enari M, et al. Oncogenic Fusion Gene CD74-NRG1 Confers Cancer Stem Cell-like Properties in Lung Cancer through a IGF2 Autocrine/Paracrine Circuit.. Cancer Res. 2016;76(4):974–983.
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi:10.1038/nature03319
- Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. doi:10.1038/nm.3074
- Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55(4):619–625. doi:10.1016/0092-8674(88)90220-6
- Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt Signaling Pathway in Mammary Gland Development and Carcinogenesis. Pathobiology. 2006;73(5):213–223.
- Zeng YA, Nusse R. Wnt Proteins Are Self-Renewal Factors for Mammary Stem Cells and Promote Their Long-Term Expansion in Culture. Cell Stem Cell. 2010;6(6):568–577. doi:10.1016/J.STEM.2010.03.020
- Yook JI, Li XY, Ota I, et al. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398–1406. doi:10.1038/ncb1508
- 42. Zhang Y, Piao B, Zhang Y, et al. Oxymatrine diminishes the side population and inhibits the expression of $β$-catenin in MCF-7 breast cancer cells. Med Oncol. 2011;28(1):99-107. doi:10.1007/s12032-010–9721-y
- DiMeo TA, Anderson K, Phadke P, et al. A Novel Lung Metastasis Signature Links Wnt Signaling with Cancer Cell Self-Renewal and Epithelial-Mesenchymal Transition in Basal-like Breast Cancer. Cancer Res. 2009;69(13):5364–5373.
- Scheel C, Eaton EN, Li SH-J, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145(6):926–940. doi:10.1016/j.cell.2011.04.029
- Sun X, Xu C, Tang SC, et al. Let-7c blocks estrogen-activated Wnt signaling in induction of self-renewal of breast cancer stem cells. Cancer Gene Ther. 2016;23(4):83–89. doi:10.1038/cgt.2016.3
- Hallett RM, Kondratyev MK, Giacomelli AO, et al. Small Molecule Antagonists of the Wnt/Beta-Catenin Signaling Pathway Target Breast Tumor-Initiating Cells in a Her2/Neu Mouse Model of Breast Cancer. PLoS One. 2012;7(3):e33976. https://doi.org/10.1371/journal.pone.0033976.
- Jang G-B, Hong I-S, Kim R-J, et al. Wnt/β-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015;75(8):1691–1702.
- Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta - Rev Cancer. 2011;1815(2):197–213. doi:10.1016/J.BBCAN.2010.12.002
- Koch U, Radtke F. Notch Signaling in Solid Tumors. Curr Top Dev Biol. 2010;92:411–455. doi:10.1016/S0070-2153(10)92013-9
- Ercan C, van Diest PJ, Vooijs M, C. Ercan, P.J. van Diest and MV. Mammary Development and Breast Cancer : The Role of Stem. Curr Mol Med. 2014;11(4):270–285.
- Yuan X, Zhang M, Wu H, et al. Expression of Notch1 Correlates with Breast Cancer Progression and Prognosis. PLoS One. 2015;10(6):e0131689.
- Zhao D, Mo Y, Li M, Zou S. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Invest. 2014;124(12):5453–5465. doi:10.1172/JCI76611.that
- Mamaeva V, Niemi R, Beck M, et al. Inhibiting Notch Activity in Breast Cancer Stem Cells by Glucose Functionalized Nanoparticles Carrying γ-secretase Inhibitors. Mol Ther. 2016;24(5):926–936. doi:10.1038/MT.2016.42
- Kondratyev M, Kreso A, Hallett RM, et al. Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene. 2012;31(1):93–103. doi:10.1038/onc.2011.212
- Zhang CC, Yan Z, Zong Q, et al. Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. 2013;2(3):233–242. doi:10.5966/sctm.2012-0096
- Harrison H, Farnie G, Howell SJ, et al. Regulation of Breast Cancer Stem Cell Activity by Signaling through the Notch4 Receptor. Cancer Res. 2010;70(2):709–718.
- McGowan PM, Simedrea C, Ribot EJ, et al. Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res. 2011;9(7):834–844.
- Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. doi:10.1158/0008-5472.CAN-06-0054
- Murone M, Rosenthal A, De Sauvage FJ. Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr Biol. 1999;9(2):76–84. doi:10.1016/S0960-9822(99)80018-9
- Denef N, Neubüser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102(4):521–531. doi:10.1016/S0092-8674(00)00056-8
- Liao X, Siu MKY, Au CWH, et al. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis. 2009;30(1):131–140. doi:10.1093/carcin/bgn230
- Wang Q, Huang S, Yang L, et al. Down-regulation of Sonic hedgehog signaling pathway activity is involved in 5-fluorouracil-induced apoptosis and motility inhibition in Hep3B cells. Acta Biochim Biophys Sin (Shanghai). 2008;40(9):819-829. doi:10.1111/j.1745–7270.2008.00456.x
- Ho J, Du Y, Wong OGW, Siu MKY, Chan KKL, Cheung ANY. Downregulation of the gli transcription factors regulator kif7 facilitates cell survival and migration of choriocarcinoma cells. PLoS One. 2014;9(9):1–8. doi:10.1371/journal.pone.0108248
- Lauth M, Toftgård R. Non-canonical activation of GLI transcription factors: Implications for targeted anti-cancer therapy. Cell Cycle. 2007;6(20):2458–2463. doi:10.4161/cc.6.20.4808
- Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–779. doi:10.1038/nature07737
- Peacock CD, Wang Q, Gesell GS, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104(10):4048–4053. doi:10.1073/pnas.0611682104
- Kasper M, Jaks V, Are A, et al. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc Natl Acad Sci U S A. 2011;108(10):4099–4104. doi:10.1073/pnas.1014489108
- Li N, Singh S, Cherukuri P, et al. Reciprocal intraepithelial interactions between TP63 and hedgehog signaling regulate quiescence and activation of progenitor elaboration by mammary stem cells. Stem Cells. 2008;26(5):1253–1264. doi:10.1634/stemcells.2007-0691
- Memmi EM, Sanarico AG, Giacobbe A, et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci. 2015;112(11):3499–3504.
- Hu K, Zhou H, Liu Y, et al. Hyaluronic acid functional amphipathic and redox-responsive polymer particles for the co-delivery of doxorubicin and cyclopamine to eradicate breast cancer cells and cancer stem cells. Nanoscale. 2015;7(18):8607–8618. doi:10.1039/C5NR01084E
- Badowska-Kozakiewicz AM, Budzik MP. Immunohistochemical characteristics of basal-like breast cancer. Wspolczesna Onkol. 2016;20(6):436–443. doi:10.5114/wo.2016.56938
- Hayes NVL, Gullick WJ. The neuregulin family of genes and their multiple splice variants in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13(2):205–214. doi:10.1007/s10911-008-9078-4
- Jeong H, Kim J, Lee Y, Seo JH, Hong SR, Kim A. Neuregulin-1 induces cancer stem cell characteristics in breast cancer cell lines. Oncol Rep. 2014;32(3):1218–1224. doi:10.3892/or.2014.3330
- 74. Kim J, Jeong H, Lee Y, Kim C, Kim H, Kim A. HRG-β1-driven ErbB3 signaling induces epithelial-mesenchymal transition in breast cancer cells. BMC Cancer. 2013;13:383. doi:10.1186/1471-2407-13-383
- Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat Rev Cancer. 2012;12(3):159–169. doi:10.1038/nrc3215
- Livingstone C. IGF2 and cancer. Endocr Relat Cancer. 2013;20(6):321–339. doi:10.1530/ERC-13-0231
- Sharma S V., Lee DY, Li B, et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell. 2010;141(1):69–80. doi:10.1016/j.cell.2010.02.027
- Osuka S, Sampetrean O, Shimizu T, et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2013;31(4):627–640. doi:10.1002/stem.1328
- Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11(11):5603–5611. doi:10.1128/MCB.11.11.5603.Updated
- Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–381. doi:10.1038/nrc3947
- Nakaoku T, Tsuta K, Ichikawa H, et al. Druggable Oncogene Fusions in Invasive Mucinous Lung Adenocarcinoma. Clin Cancer Res. 2014;20(12):3087–3093.
- Fernandez-Cuesta L, Plenker D, Osada H, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014;4(4):415–422.
- He J, Sheng T, Stelter AA, et al. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J Biol Chem. 2006;281(47):35598–35602. doi:10.1074/jbc.C600200200
- Schneider FT, Schänzer A, Czupalla CJ, et al. Sonic hedgehog acts as a negative regulator of β-catenin signaling in the adult tongue epithelium. Am J Pathol. 2010;177(1):404–414. doi:10.2353/ajpath.2010.091079